Phenylketonurie (PKU) ist eine angeborene Stoffwechselstörung, die auf einer fehlenden oder verminderten Aktivität der Phenylalaninhydroxylase beruht, dem Enzym, das für die Umwandlung von Phenylalanin zu Tyrosin verantwortlich ist. Das nicht abgebaute Phenylalanin (Phe) reichert sich im Blut und in anderen Körpergeweben an. Über alternative Stoffwechselwege wird Phe zum Teil in Phenylketone umgewandelt. Die hohen Phenylalanin- und Phenylketonspiegel und die einhergehenden niedrigen Tyrosinspiegel führen zu Gedeihstörungen, neurologischen Schäden, geistiger Beeinträchtigung und Epilepsie.
Genetisch betrachtet ist die PKU eine autosomal-rezessiv vererbte Störung. Das heißt, die betroffene Person besitzt zwei PKU-Gene, eines von jedem Elternteil.
Abhängig vom Ursprungsland betrifft die PKU zwischen 1:10.000 und 1:20.000 Menschen. In einigen Ländern ist die Prävalenz höher. So erreicht die Geburtenprävalenz in der Türkei eine Rate von 1:2.600.
Die PKU wird im Rahmen des Neugeborenenscreenings erfasst. Das Screening von Neugeborenen auf PKU ist heute in vielen Ländern vorgeschrieben. Eine frühzeitige Diagnose und Behandlung sind extrem wichtig. Unbehandelt kann die PKU zu einer schweren geistigen Retardierung führen. Bei sofortiger Einleitung einer sorgfältigen Behandlung ist eine normale Entwicklung möglich, und Patienten mit PKU können ein gesundes Leben führen.
Die diätetische Therapie basiert auf einer Restriktion der Proteinzufuhr und Supplementation mit einer speziellen Aminosäurenmischung, um die Phe-Spiegel im Blut auf einem sicheren Niveau zu halten.
Nahrungsmittel und Getränke mit einem hohen Gehalt an Phe sollten vermieden werden. Nahrungsmittel und Getränke mit niedrigeren Phe-Konzentrationen können in Maßen verzehrt werden.
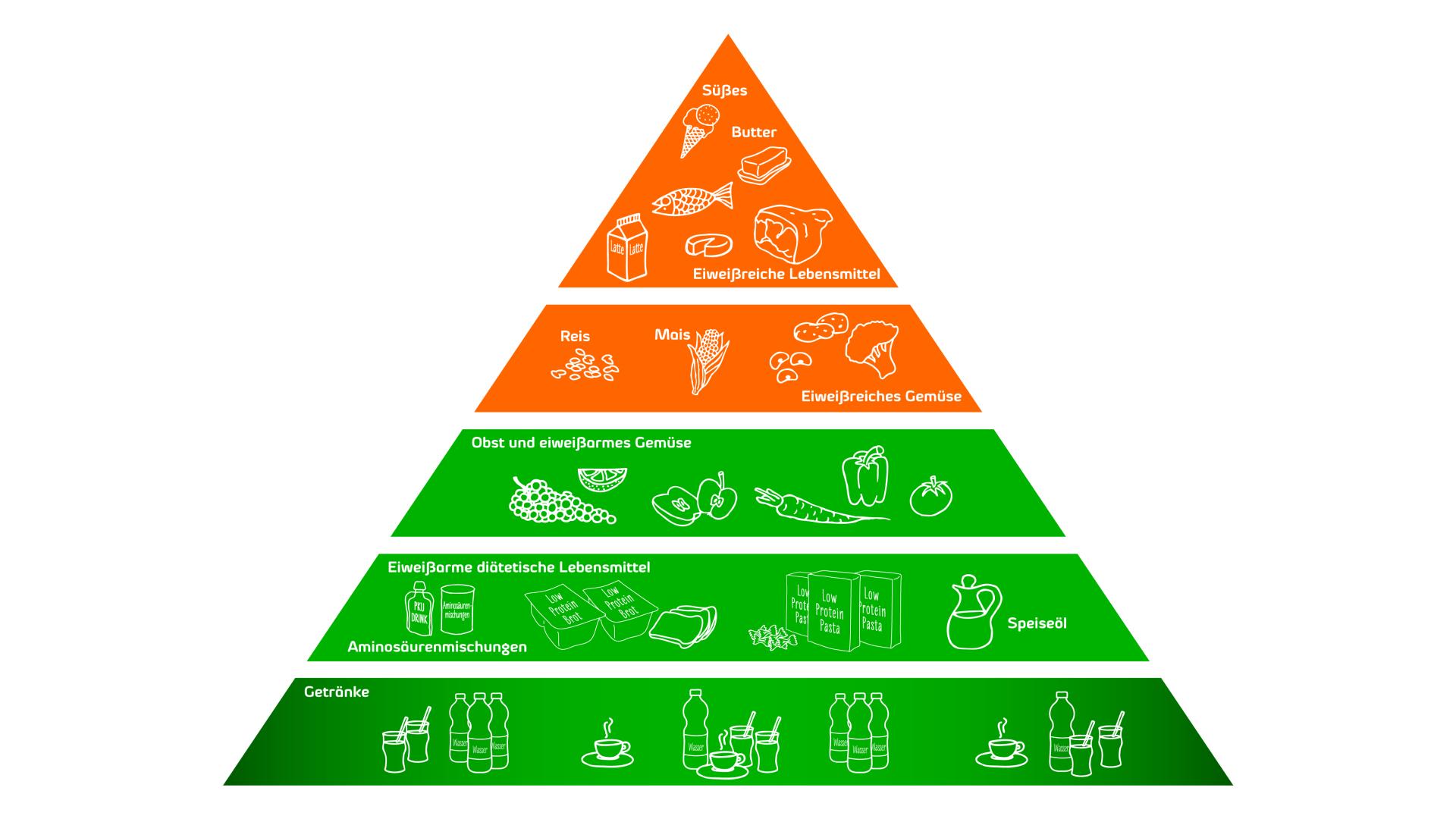
Da Kinder und insbesondere Säuglinge für ein normales Wachstum und eine altersgerechte Entwicklung eine ausreichende Proteinzufuhr benötigen, wurden spezielle Formulierungen entwickelt, um die benötigten proteinreichen Nahrungsmittel zu ersetzen. Für Säuglinge hat Comidamed die Milchersatznahrung comida-PKU A formula, eine phenylalaninfreie Aminosäurenmischung speziell für Säuglinge im ersten Lebensjahr entwickelt. Als Spezialnahrung für Kinder ab 1 Jahr, Jugendliche und Erwachsene ist comida-PKU B formula und comida-PKU B erhältlich. comida-PKU C ist eine konzentrierte phenylalaninfreie Aminosäurenmischung für Jugendliche und Erwachsene ab 15 Jahren.
