La fenilcetonuria es un trastorno metabólico hereditario provocado por un defecto en la enzima fenilalanina hidroxilasa, encargada de convertir el aminoácido fenilalanina en tirosina. Como el cuerpo no es capaz de transformar ni de eliminar la fenilalanina (Phe), este aminoácido se acumula en la sangre y otros tejidos corporales. En algunos casos transforma la fenilalanina en una sustancia llamada fenilcetona. El elevado nivel de fenilalanina y fenilcetona y los bajo niveles de tirosina pueden provocar problemas de crecimiento, daños neurológicos, discapacidad intelectual y epilepsia.
En términos genéticos, la fenilcetonuria es un trastorno autosómico recesivo, es decir, la persona afectada tiene dos genes de fenilcetonuria, uno procedente de cada progenitor.
La prevalencia de la fenilcetonuria oscila entre 1 de cada 10000 a 1 de cada 20000 personas y puede variar significativamente en función del país. En Turquía, por ejemplo, uno de cada 2600 recién nacidos tiene fenilcetonuria.
La fenilcetonuria se diagnostica a través de un pequeño análisis de sangre que se realiza a los recién nacidos. En muchos países esta prueba está dentro de un plan de salud y es obligatoria. La detección precoz y el tratamiento son de vital importancia ya que, sin un tratamiento adecuado, la persona puede sufrir daños irreversibles. Sin embargo, si se inicia en seguida con el tratamiento, las personas afectadas pueden llevar una vida sana y normal.
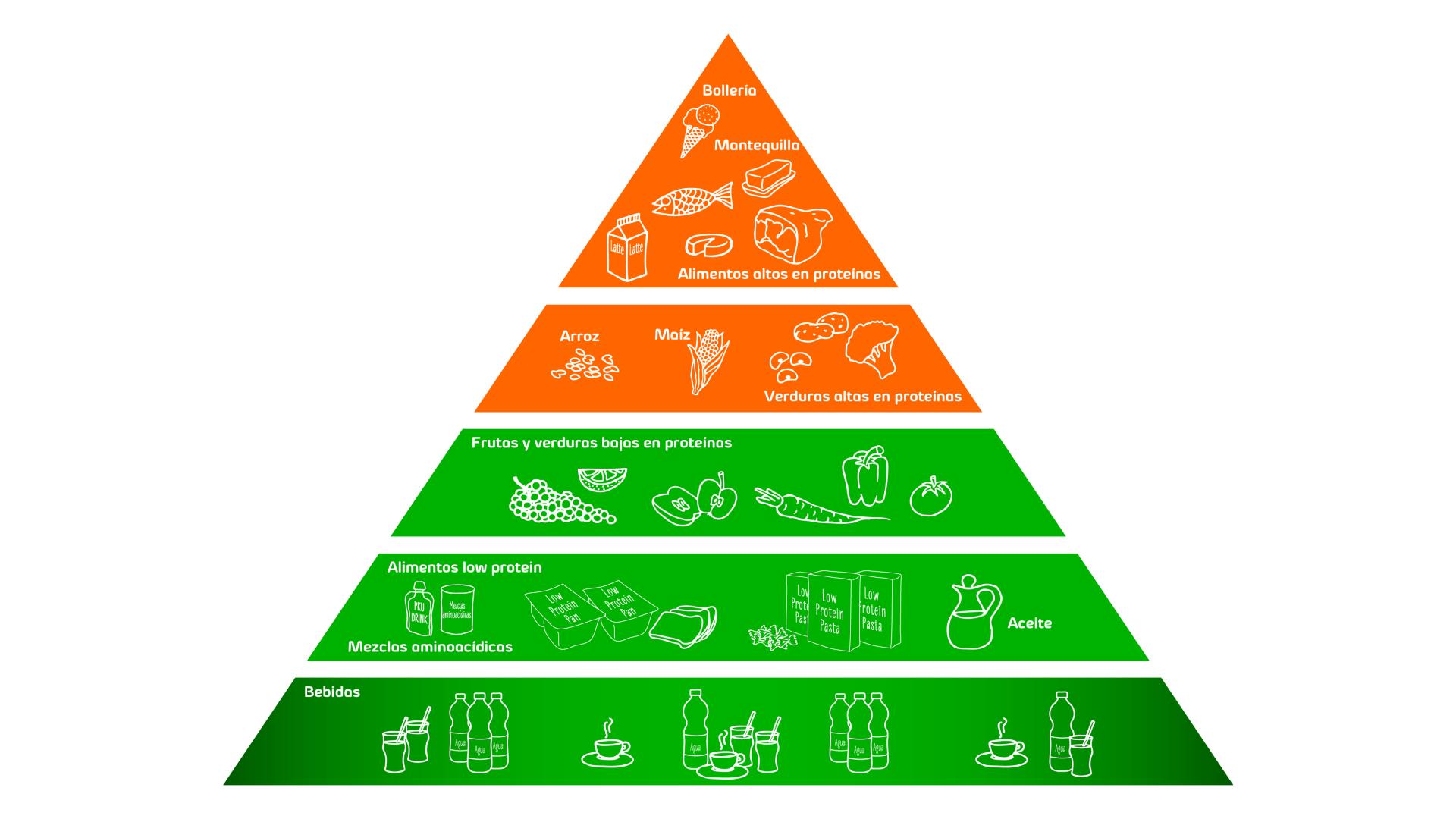
El tratamiento de la fenilcetonuria consiste en una dieta especial que restringe la ingesta de proteína natural –que contiene fenilalanina – y sustituye los aminoácidos esenciales para el cuerpo con fórmulas especiales. El objetivo de esta terapia es mantener los niveles de fenilalanina dentro de un rango de tolerancia.
Las personas con fenilcetonuria deben evitar comidas y bebidas que contienen elevados niveles de fenilalanina y deben basar su alimentación en productos bajos in fenilalanina. También deben evitar bebidas o comidas endulzadas con aspartamo al ser este fuente de fenilalanina.
Comidamed ofrece una gama de productos para la gestión nutricional de la fenilcetonuria. Estos suplementos están especialmente desarrollados para diferentes grupos de edad. El producto PKUmed A formula por ejemplo es una fórmula completa que aparte de la mezcla de aminoácidos libres de fenilalanina también aporta todos los otros nutrientes necesarios para el desarrollo sano de un lactante. A partir de un año los pacientes disponen de diferentes productos con mayor o menor aporte calórico. Será función del médico o nutricionista experto de recomendar el producto más adecuado a la situación particular de cada paciente.

