Фенилкетонурия (ФКУ)
The following information provides a general overview of Phenylketonuria and is intended for educational purposes only. It should not replace guidance from qualified healthcare professionals. Vitaflo™ International Limited accepts no responsibility for any loss resulting from reliance on the content. For comprehensive and up-to-date recommendations, please refer to national and international clinical guidelines.
Что такое Фенилкетонурия (comida-PKU)?
Фенилкетонурия (ФКУ) - наследственное, аутосомно-рецессивное заболевание ведущее к нарушению белкового обмена. Это означает:
Что является причиной Фенилкетонурии?
Фенилкетонурия (ФКУ) является следствием дефицита печеночного фермента: фенилаланингидроксилазы.
Ферменты подобны химическим ножницам и необходимы организму для расщепления белка на аминокислоты. Белок состоит из 20 аминокислот.
Некоторые из аминокислот являются незаменимыми, они не могут вырабатываться в организме естественным путем, поэтому они должны поступать с пищей.
Фенилаланин является одной из таких незаменимых аминокислот.
Фенилаланингидроксилаза отвечает за преобразование аминокислоты фенилаланин в аминокислоту тирозин. Организм использует эти аминокислоты для роста и развития всех органов и систем организма, а также выработки нейромедиаторов необходимых для работы мозга и меланина (пигмента в коже).
Ниже можно рассмотреть схематическое изображение:
*Note: you may find our Phenylketonuria (PKU) condition-specific resource helpful. This is intended for use with patients and families to explain the complex science behind the inheritance of PKU, diagnosis and management.
You can also direct your patients onto this site to read the same information.
Как проявляется недостаточность фермента при ФКУ?
При накоплении фенилаланина
Он становится токсичным и вызывает необратимые повреждения головного мозга, которые приводят к нарушениям способности к обучению, поведенческими изменения, тремору (дрожание конечностей) и судорогам.
Низкий уровень тирозина
При отсутствии лечения низкий уровень тирозина приводит к нарушению выработки нейромедиаторов и дефициту меланина, сопровождающемуся характерной бледностью кожи.
Выявление Фенилкетонурии: скрининг новорожденных
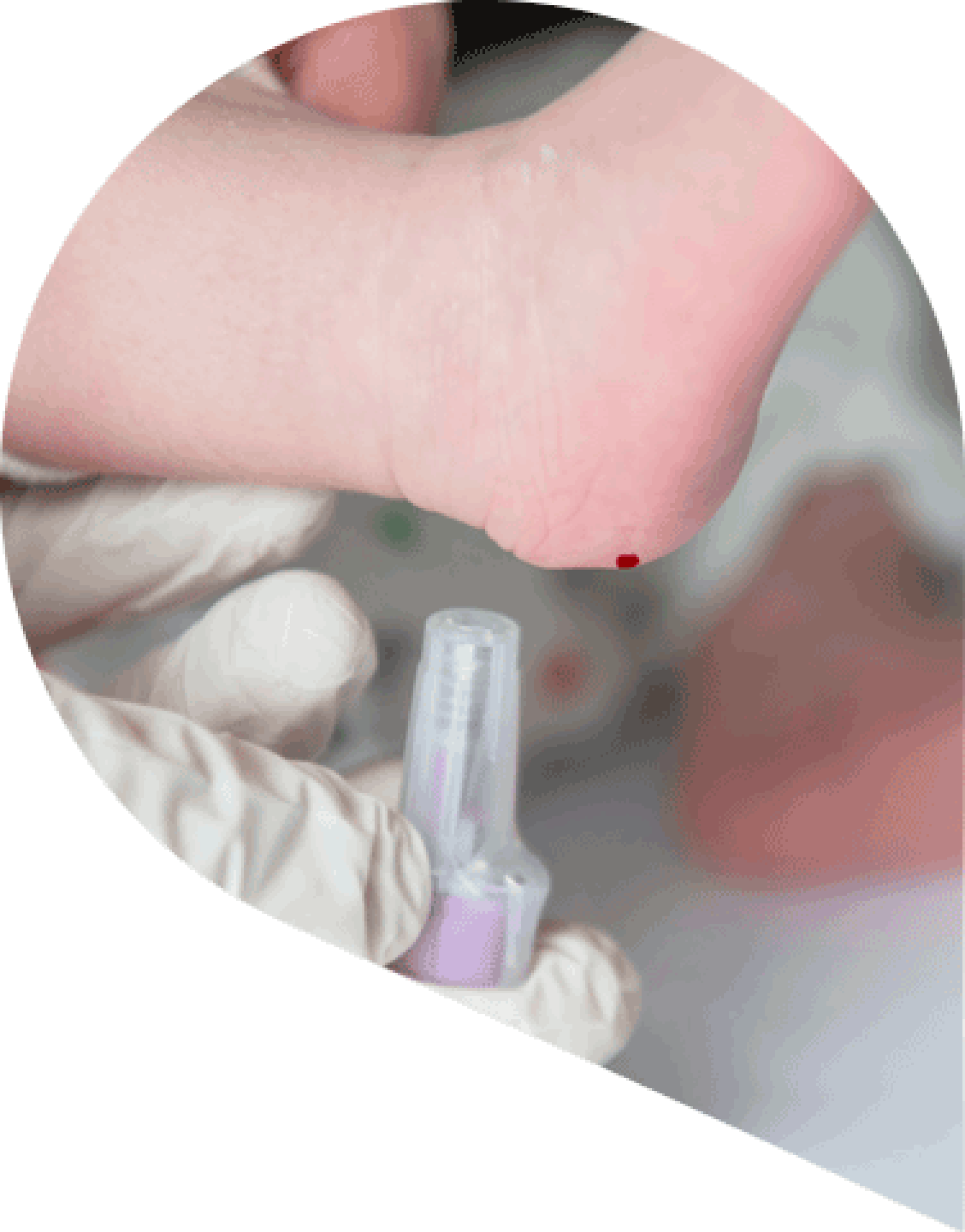
Почти во всех случаях фенилкетонурия диагностируется с помощью неонатального скрининга новорожденных (НСН).
Для проведения неонатального скрининга медицинский работник через несколько дней после рождения ребенка берет образец крови из пяточки и отправляет в лабораторию для анализа уровня фенилаланина.
При получении положительного результата с родителями или опекунами незамедлительно связываются для дальнейшего обследования и подтверждения диагноза.
Насколько распространена фенилкетонурия?
Фенилкетонурия - редкое заболевание, однако это наиболее распространенное врожденное нарушение белкового обмена.
Мировой показатель распространенности заболевания составляет 1 из 10 000 - 12 0001,2. Это означает, что во всем мире 1 из 12 000 новорожденных рождается с Фенилкетонурией.
Каковы симптомы Фенилкетонурии?
Если не лечить фенилкетонурию она может проявляться по-разному:
- Затхлый запах тела
- Необратимые повреждения мозга
- Бледность кожи лица
- Тремор
- Судороги
- Проблемы с кожей, такие как экзема
- Трудности в обучении
- Проблемы с поведением
Однако, если пациентам своевременно поставили диагноз и назначили специальную диету на ранней стадии, то эти симптомы можно предотвратить, а ожидаемая продолжительность жизни будет такой же, как у здорового населения.
Как лечат Фенилкетонурию в раннем детском возрасте?
Лечение фенилкетонурии (ФКУ) состоит из 4 составляющих:
- Исключение из диеты продуктов с высоким содержанием белка
- Учет доли фенилаланина (ФА) в диете в соответствии с установленной индивидуальной переносимостью
- Включение в рацион продуктов с низким содержанием белка
- Обязательное употребление специализированных аминокислотных смесей без фенилаланина для замены белка
Исключение из диеты продуктов с высоким содержанием белка
Высокий уровень фенилаланина (ФА) содержится во многих продуктах, являющихся источником белка, таких как:
- Мясо
- Рыба
- Яйца
- Орехи
- Молоко и молочные продукты
- Соя
- И другие пищевые продукты
Чтобы поддерживать уровень ФА под контролем, больным ФКУ важно ограничить потребление белка, отказываясь от продуктов с высоким содержанием белка.

Examples of high protein foods
Учет количества фенилаланина (ФА) в диете в соответствии с установленной индивидуальной переносимостью
Небольшое строго контралируемое количество феналиланина должно быть включено в рацион питания для предотвращения дефицита фенилаланина.
Количество ФА, которое пациент с ФКУ может безопасно употреблять без токсических проявлений, называется «индивидуальной переносимостью ФА» или «толерантностью к ФА». Это значение может варьироваться индивидуально у каждого пациента.
Система, используемая для обеспечения достаточного потребления ФА в рационе без превышения допустимых значений, варьируется в зависимости от страны.
Различные системы помогают классифицировать продукты и рассчитывать пищевой рацион в соответствии с содержанием ФА (например, проверять информацию о пищевой ценности или составе); медицинские работники могут оказать поддержку в этом, и их рекомендации будут основаны на отказе от продуктов с высоким содержанием белка (как источника ФА), и равномерном распределение потребления допустимых количеств ФА в течение дня и включении достаточного количества низкобелковых продуктов в рацион.
Продукты с низким содержанием белка
Наряду с исключением из рациона продуктов с высоким содержанием белка, при диетотерапии ФКУ в равной степени важно включать в рацион продукты с низким содержанием белка.
Это могут быть продукты с естественным низким содержанием белка, например - большинство фруктов и некоторые овощи, жиры, масла и многие продукты с высоким содержанием сахара; или же это могут быть специализированные низкобелковые продукты промышленного производства, такие как: низкобелковый хлеб, заменители молока, мучные смеси, макароны и рис.
В ряде стран специализированные низкобелковые продукты можно получить по программам льготного обеспечения. Такие продукты часто содержат большое количество калорий, что помогает пациентам удовлетворять энергетические потребности.
Также, низкобелковые продукты могут обеспечить большую схожесть с рационом питания здорового человека, позволяя разнообразить блюда в течение недели.

Аминокислотные смеси для замены белка
Хотя естественная переносимость белка при фенилкетонурии (ФКУ) варьируется, общая потребность в белке в сутки при ФКУ аналогична людям без ФКУ.
Общая потребность в белке в сутки не может быть удовлетворена за счет небольшого количества натурального белка в рационе пациентов с ФКУ. Чтобы обеспечить достаточное потребление белка при ФКУ, используют специализированные смеси на основе аминокислот без фенилаланина, или сокращенно «заменители белка».
Заменители белка для пациентов с ФКУ обеспечивают "белковый эквивалент" – они содержат все аминокислоты, которые есть в натуральном белке, за исключением фенилаланина. В таких специализированных продуктах фенилаланин, как правило, полностью отсутствует, а в некоторых странах в ассортименте доступны смеси с небольшим количеством фенилаланина в составе.
Как правило, большинство пациентов принимают одну порцию аминокислотной смеси за завтраком, одну во второй половине дня и одну вечером перед сном, чтобы равномерно распределить потребление в течение дня.
How is Phenylketonuria managed in infancy?
Dietary management is ideally started as early as possible following diagnosis, to reduce blood phenylalanine (phe) levels to target ranges as quickly as possible1-2.
This will involve balancing intakes of breast milk or standard infant formula (which provides phe), with a phe free formula for infants, to achieve good metabolic control, meet nutritional requirements and optimise growth1,6.
Breastfeeding offers many benefits to mothers and infants, and a family should be supported in making the appropriate choice for their family, if deemed appropriate by the metabolic healthcare team.
A specialist metabolic team should be responsible for the management of the individual with PKU from diagnosis and throughout life.
You may find the following resource helpful.


Information presented on this page is intended for healthcare professional use only. Food for Special Medical Purposes to be used under medical supervision.
Date of publishing: 10/4/2025
- van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Beblo S, Blau N, et al. European guidelines on diagnosis and treatment of phenylketonuria: first revision. Mol Genet Metab. 2025;144:109125. https://doi.org/10.1016/j.ymgme.2025.109125
- Smith WE, Berry SA, Bloom K, Brown C, Burton BK, Demarest OM, et al. Phenylalanine hydroxylase deficiency diagnosis and management: a 2023 evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2025;27:101289. https://doi.org/10.1016/j.gim.2024.101289
- van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12:162. https://doi.org/10.1186/s13023-017-0685-2
- Hillert A, Anikster Y, Bélanger-Quintana A, Burlina A, Burton BK, Carducci C, et al. The genetic landscape and epidemiology of phenylketonuria. Am J Hum Genet. 2020;107:234–50. https://doi.org/10.1016/j.ajhg.2020.06.006
- Singh RH, Rohr F, Frazier D, Cunningham A, Mofidi S, Ogata B, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. 2014;16:121–31. https://doi.org/10.1038/gim.2013.179
- MacDonald A, van Wegberg AMJ, Ahring K, Beblo S, Bélanger-Quintana A, Burlina A, et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis. 2020;15:171. https://doi.org/10.1186/s13023-020-01391-y
