Hereditary Tyrosinaemia type I (HT1)
The following information provides a general overview of Hereditary Tyrosinaemia type I and is intended for educational purposes only. It should not replace guidance from qualified healthcare professionals. Vitaflo™ International Limited accepts no responsibility for any loss resulting from reliance on the content. For comprehensive and up-to-date recommendations, please refer to national and international clinical guidelines.
What is Hereditary Tyrosinaemia?
Hereditary Tyrosinaemia (HT, can also be spelt ‘tyrosinemia) is a group of recessively inherited disorders of tyrosine (tyr) metabolism, resulting in the accumulation of tyr and its metabolites in the blood.
There are three types of HT1
HT1 is the most common form, which causes a build-up of toxic metabolites in both the blood and urine and can be life-threatening.
Raised tyr levels can also occur due to other causes, such as preterm infants and severe liver disease. In fact, around 1 in 10 of all newborns have temporarily elevated levels of tyr (transient Tyrosinaemia), but these cases are not genetic and are more likely due to vitamin C deficiency or an immature liver due to premature birth.
The following information on this webpage refers to HT1 only.
What is the cause of Tyrosinaemia?
Hereditary Tyrosinaemia type I (HT1) is characterised by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH), involved in the final step of tyrosine (tyr) breakdown2.
This leads to raised levels of maleylacetoacetate (MAA), fumarylacetoacetate (FAA), and succinylacetone (SA)3. These compounds are toxic and cause the symptoms of HT1.
*Note: you may find our condition-specific resource helpful. This is intended for use with patients and families to explain the complex science behind the inheritance of Tyrosinaemia, diagnosis and management.
For further information on resources available, please enquire: Contact page
What are the symptoms of Tyrosinaemia?
Hereditary Tyrosinaemia type I (HT1) symptoms and severity can vary and are usually classified according to age of onset of symptoms and severity2-3.
Typically presents before an infant is 6 months of age with acute liver failure. The most severe cases tend to present before two months of age.
Often present within the first year of life with progressive liver failure, enlargement of the liver and spleen, coagulopathy (inability of blood to clot), faltering growth and rickets present.
Can present at any age over the age of one year, with liver and/or renal disease, rickets and neurological issues due to porphyria, cardiomyopathy or pancreatic cell hypertrophy.
If left untreated, HT1 can lead to hepatic cancer and ultimately, childhood death. Thankfully, treatment options are available.
Testing for Tyrosinaemia: newborn screening
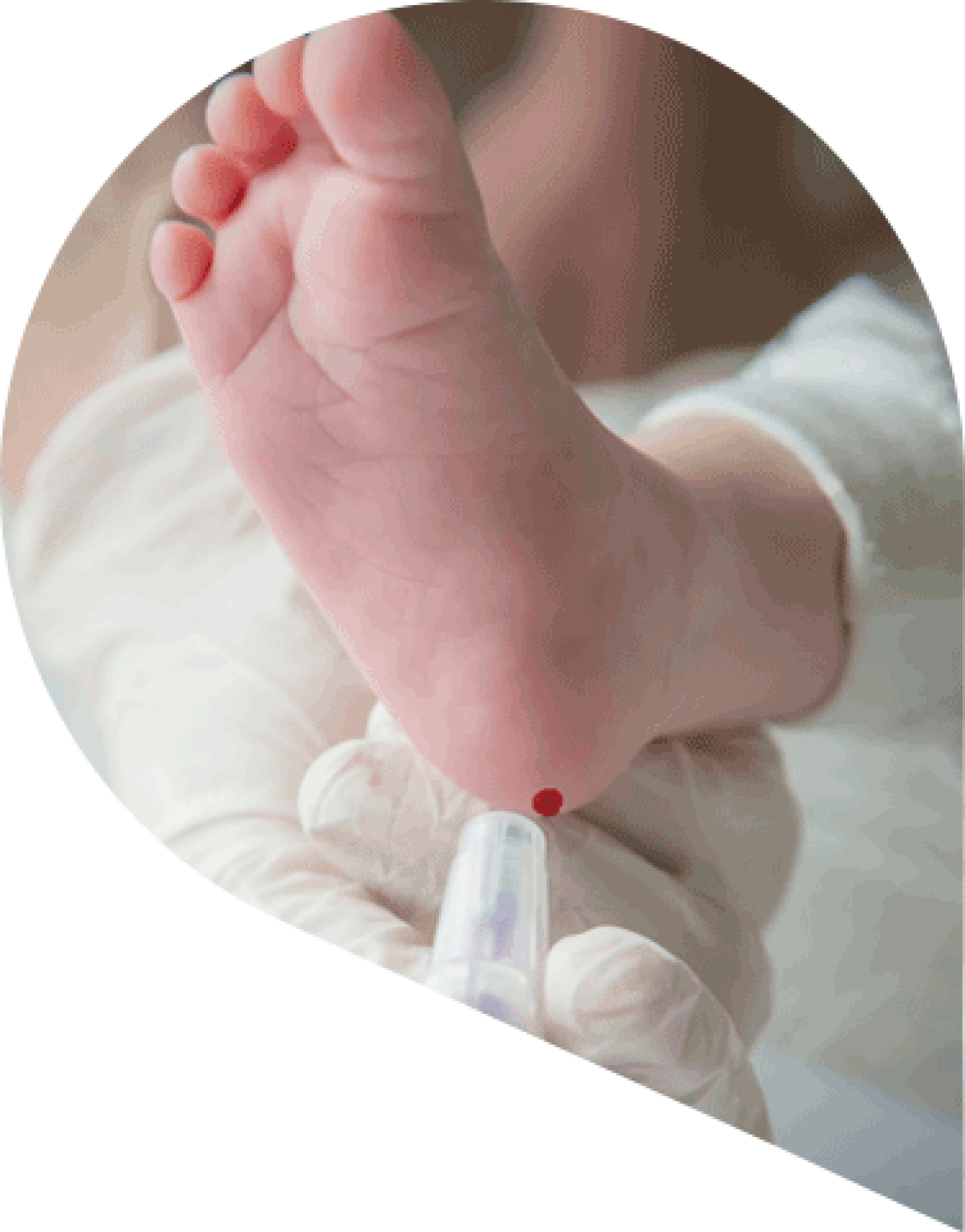
Hereditary Tyrosinaemia type I (HT1) can be detected via newborn screening (NBS), however it is not widely available due to the rarity of the condition3.
The presence of succinylacetone (SA) is indicative of HT1, and further investigation and immediate management are required2-3.
How common is Tyrosinaemia?
Hereditary Tyrosinaemia type I (HT1) is a rare condition with a global incidence rate of 1 in 100,000, but the most common mutation is found in the French-Canadian population, which has an incidence of 1 in 8,0001-2,4.
How is Tyrosinaemia managed?
Hereditary Tyrosinaemia type I (HT1) is managed through the life-long use of the drug Nitisinone (NTBC) alongside a low protein diet2-3.
Whilst NTBC prevents the accumulation of succinylacetone (SA), it does lead to a secondary increase in blood tyrosine (tyr) levels5. Therefore, dietary management is key to help lower tyr levels.
The dietary management of HT1 consists of 4 different components:
- Avoidance of high protein foods
- A measured amount of phenylalanine (phe) and tyr from food according to patient tolerance
- Low-protein foods
- Protein substitutes
Avoidance of high-protein foods

Example of high protein foods.
High levels of phe and tyr are present in many foods that contain protein, such as:
- meats
- fish
- eggs
- nuts
- milk
- dairy products
- soya
- and many more.
To help keep tyr levels within target range2-3, a low protein diet is required and avoidance of high protein foods.
A measured amount of tyr and phe according to patient tolerance
As phe is an essential amino acid, a small, measured amount of phe must be included in the diet to prevent deficiency3,6.
The amount of tyr and phe that an individual with HT1 can safely consume without the tyr building to toxic levels is known as protein tolerance and will vary from individual to individual.
The system used to ensure an adequate tyr and phe intake, without exceeding tyr and phe tolerance, varies from country to country. Often, a phe or protein exchange system is used, which helps to categorise and count foods according to their phe content (such as checking the nutritional information or the ingredients).
Low protein foods
As well as avoiding high protein foods, incorporating low protein foods into the diet is equally essential for the dietary management of HT1.
They can be foods naturally low protein, such as most fruits and some vegetables, fats, oils and many high-sugar foods. Specially manufactured foods, such as bread, milk alternatives, flour mixes, pasta and rice, subject to availability, can also be used.
These types of foods can help provide increased variety, energy, and a sense of normality to patients’ diets.

Protein substitutes
Although natural protein tolerance varies in HT1, total protein requirements are like those of individuals without HT1.
Total protein requirements will not be met by the small amount of natural protein that HT1 patients would consume through their diet.
Therefore, to help achieve adequate protein intake in HT1, products called protein substitutes are essential. They provide a suitable phe and tyr free protein source and ensure that all other essential amino acids and nutrients are supplied to support normal protein synthesis for growth and development.
Typically, most patients with HT1 consume their protein substitute with meals, spread evenly throughout the day.
How is Tyrosinaemia managed in infancy?
The management of Hereditary Tyrosinaemia type I (HT1) in infancy will depend on the presenting tyrosine (tyr) and succinylacetone levels (SA).
For all infants with raised tyr levels, Nitisinone (NTBC) will be introduced immediately in conjunction with a phenylalanine (phe) and tyr free protein substitute. However, with very high tyr levels some infants may have to temporarily stop their intake of breast milk or standard infant formula until tyr levels fall6.
Once tyr levels have reduced to an acceptable level, breast milk or standard infant formula can be reintroduced to provide essential phe6. Breastfeeding offers many benefits to mothers and infants and should be supported wherever possible.
A specialist metabolic team should be responsible for the management of the individual with HT1 from diagnosis and throughout life.
You may find following resource helpful:

- Chakrapani A, Gissen P, McKiernan P. Disorders of tyrosine metabolism. In: Saudubray J-M, van den Berghe G, Walter JH, editors. Inborn metabolic diseases: diagnosis and treatment. 6th ed. Berlin: Springer; 2022. p. 355–67. https://doi.org/10.1007/978-3-662-63123-2_17
- De Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;8:8. https://doi.org/10.1186/1750-1172-8-8
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19:1380–95. https://doi.org/10.1038/gim.2017.101
- Morrow G, Tanguay RM. Biochemical and clinical aspects of hereditary tyrosinemia type 1. In: Mandel H, Tanguay RM, editors. Hereditary tyrosinemia. Adv Exp Med Biol. 2017;959:9–21. https://doi.org/10.1007/978-3-319-55780-9_2
- Introne WJ. Alkaptonuria. 2016. https://doi.org/10.1093/med/9780199972135.003.0015
- Dixon M, MacDonald A, White F. Disorders of amino acid metabolism, organic acidaemias and urea cycle disorders. In: Shaw V, editor. Clinical paediatric dietetics. 5th ed. Hoboken (NJ): Wiley-Blackwell; 2020. p. 513–98.
Related products
Browse the products available for managing this condition.


Information presented on this page is intended for healthcare professional use only. Food for Special Medical Purposes to be used under medical supervision.
Date of publication: 30/05/2025
