Homocystinuria (HCU)
The following information provides a general overview of Homocystinuria and is intended for educational purposes only. It should not replace guidance from qualified healthcare professionals. Vitaflo™ International Limited accepts no responsibility for any loss resulting from reliance on the content. For comprehensive and up-to-date recommendations, please refer to national and international clinical guidelines.
What is Homocystinuria?
Homocystinuria (HCU) is a group of inherited, autosomal, recessive disorders of homocysteine (amino acid) metabolism1. Homocysteine forms during the catabolism of methionine (essential amino acid), resulting in the accumulation of homocysteine, methionine and other sulphur-containing metabolites and a relative deficiency of cysteine in the blood1.
What is the cause of Homocystinuria?
Homocystinuria (HCU) is caused by a deficiency of the enzyme cystathionine β-synthase (CβS)1. CβS converts homocysteine to cystathionine, which is in turn converted to cysteine, a non-essential amino acid. However, in HCU, cysteine becomes conditionally essential.
There are two main phenotypes of HCU1:
- Pyridoxine-responsive HCU
- Pyridoxine-non-responsive HCU, also known as ‘Homocystinuria’ or Classical HCU
As CβS is a pyridoxine (vitamin B6, used as a co-factor) dependent enzyme, any newly diagnosed individuals must be assessed for their responsiveness to pharmacological doses of pyridoxine.
Although individuals with HCU are not deficient in pyridoxine, providing pharmacological doses of pyridoxine to some individuals with HCU can effectively manage their condition.
Others with HCU are not responsive, or only partly responsive, to these large doses of pyridoxine. This means they must have their condition managed by other means.
The following information on this webpage refers to Classical HCU only.
*Note: you may find our condition-specific resource helpful, intended for use with patients and families to explain the complex science behind the inheritance of homocystinuria, diagnosis and management.
You can also direct your patients to this site to read the same information.
For further information on resources available, please enquire: Contact page
What are the symptoms of Homocystinuria?
Classical homocystinuria (HCU) can present with a wide spectrum of symptoms, at any age from infancy through to adulthood.
Individuals who have HCU present clinically normal at birth; thus, early diagnosis (if no newborn screening is in place) and management are crucial to prevent the progressive multisystem clinical features of HCU.
HCU is associated with abnormalities in four organs/ organ systems. It can involve all four systems or only one1-3:
Without treatment, dislocation of the ocular lens (ectopia lentis), myopia, and glaucoma are frequent, severe, and characteristic complications of HCU. These generally occur during childhood.
Patients with HCU tend to be tall, with bone elongation (abnormally tall stature with long arms and legs) and prone to osteoporosis, highly arched feet, knock knees, curved spine (scoliosis), and increased risk of broken bones.
Developmental delay and mental retardation affect over half patients to a variable degree of severity. Seizures and psychiatric disturbances have also been reported.
Thromboembolism is the major cause of morbidity and mortality in individuals with HCU. Thrombophlebitis and pulmonary embolism are the most common vascular incidents.
Children are more likely to present with developmental delay and lens dislocation. Adults are more likely to be diagnosed following vascular events. However, most manifestations can occur at almost any age1.
If left untreated, seizures, psychiatric disorders, and thromboembolic events (such as cerebral ischemia, myocardial infarction, and pulmonary embolism) may occur1.
However, the risk of these long-term complications can be reduced with lifelong management of HCU and regular monitoring.
Testing for Homocystinuria: newborn screening
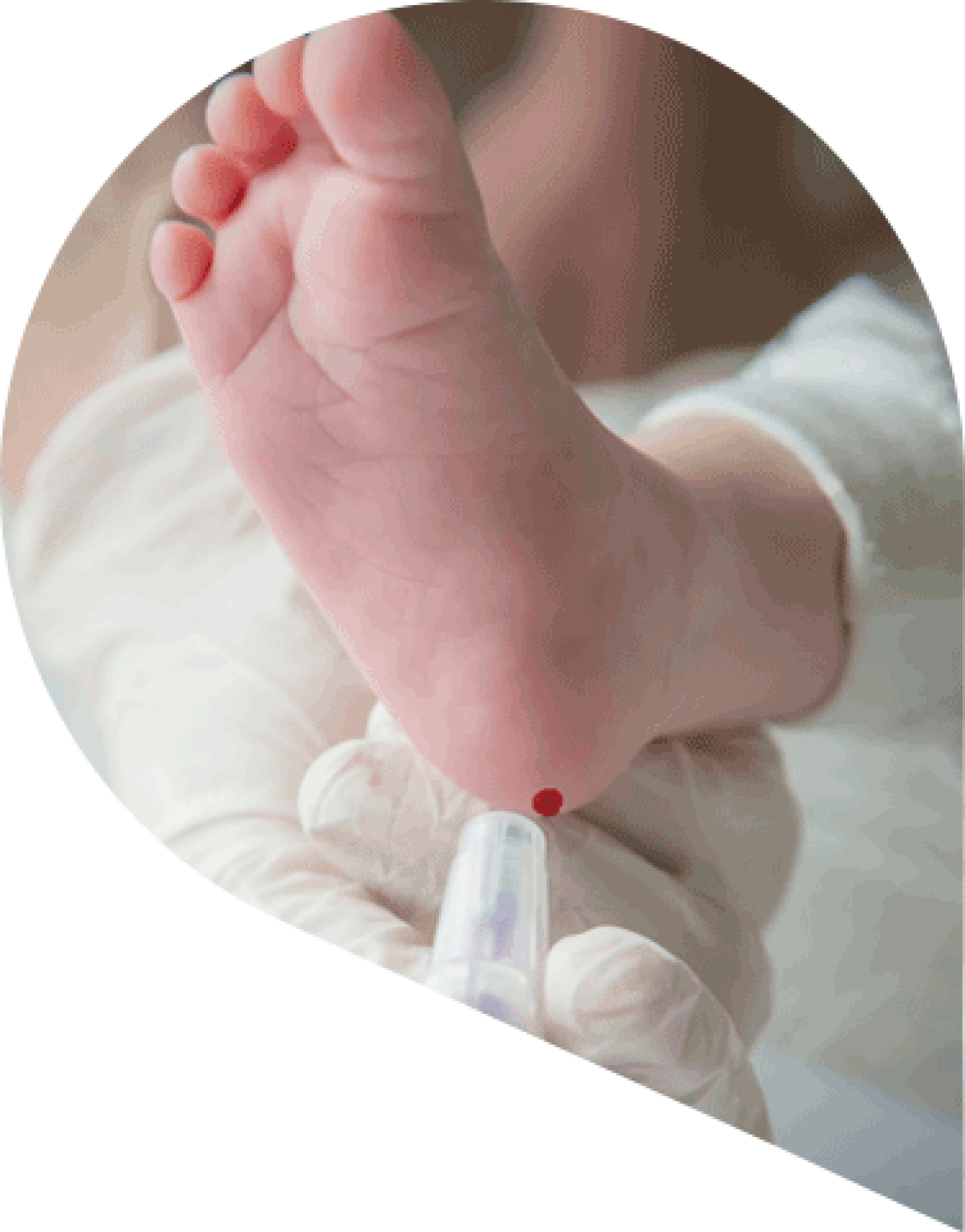
Classical homocystinuria (HCU) can be diagnosed at birth via newborn screening (NBS) however, not all countries screen for HCU1.
Elevated plasma methionine and total homocysteine (tHcy) (typically > 100 μmol/L) are used to diagnose HCU1-2.
Pyridoxine responsiveness is also assessed during the NBS diagnosis with a trial of a pharmacological dose of pyridoxine (vitamin B6) for 7-14 days, in those diagnosed shortly after birth.
Individuals who are not diagnosed with NBS will typically remain undiagnosed until they become symptomatic; some children may be picked up due to their physical stature or eye issues, whereas some adults may present with a cardiovascular event4.
How common is Homocystinuria?
The worldwide population prevalence for Homocystinuria (HCU) varies.
The global prevalence is 1 in 344,000 and is higher in countries such as Ireland (1 in 65,000) and Qatar (1 in 1800)1,5.
How is homocystinuria managed?
The primary aim of management in Classical homocystinuria (HCU, i.e. pyridoxine non-responsive) is to achieve good metabolic control by reducing blood methionine and total homocysteine (tHcy) levels to within recommended target levels (<100 µmol/L)1.
This is achieved through a low methionine diet in conjunction with medications if target homocysteine (Hcy) levels are not achieved1.
Betaine is a drug that is used to lower plasma Hcy levels by serving as a methyl donor for the re-methylation of Hcy to generate methionine1-2.
There are five main components of the dietary management of HCU1-2:
- Avoidance of high protein foods
- A measured amount of methionine from foods titrated to patients’ biochemistry
- Low protein foods
- Protein substitutes
- Supplementation (if required) of cysteine, vitamin B12 and folate
Avoidance of high-protein foods
High levels of methionine are present in many foods that contain protein, such as:
- meats
- fish
- eggs
- nuts
- milk
- dairy products
- soya
- and many more.
To help keep blood methionine and homocysteine (Hcy) levels within target, it is important that protein intake is restricted, which means avoiding high-protein foods.

Example of high protein foods.
A measured amount of methionine from foods according to patient tolerance
As methionine is an essential amino acid, a small, measured amount must be included in the diet to prevent methionine deficiency1.
The amount of methionine that an individual with Classical homocystinuria (HCU) can safely consume, whilst achieving target blood homocysteine (Hcy) and methionine levels is known as protein tolerance, and will vary from individual to individual.
The system used to ensure an adequate methionine intake, without exceeding tolerance, varies from country to country. An example is a methionine exchange system. This helps to categorise and count foods according to methionine content (such as checking the nutritional information or the ingredients).
Low protein foods

As well as avoiding high protein foods, incorporating low protein foods into the diet is equally essential for the dietary management of Classical homocystinuria (HCU).
They can be foods naturally low protein, such as most fruits and some vegetables, fats, oils and many high-sugar foods. Specially manufactured foods, such as bread, milk alternatives, flour mixes, pasta and rice, subject to availability, can also be used.
These types of foods can help provide increased variety, energy, and a sense of normality to patients’ diets.
Protein substitutes
Although natural protein tolerance varies in patients with Classical homocystinuria (HCU), total protein requirements are like those of individuals without it.
Total protein requirements will not be met by the small amount of natural protein that HCU patients would get in their diet.
Therefore, to help achieve adequate protein intakes in HCU, products called protein substitutes are essential. They provide a suitable methionine free protein source and ensure all other essential amino acids and nutrients are supplied to support normal protein synthesis for growth and development1-2.
Typically, most HCU patients consume their protein substitute with meals, spread evenly throughout the day.
Cysteine supplements, along with vitamin B12 and folate may be required to correct any deficiencies1.
How is Homocystinuria managed in infancy?
Dietary management is ideally started as early as possible, following diagnosis to reduce plasma homocysteine (Hcy) and methionine to target ranges, as quickly as possible. This will involve balancing intakes of breast milk or standard infant formula (which provide methionine) with a methionine-free formula for infants, to achieve good metabolic control and optimised growth.
Breastfeeding offers many benefits to mothers and babies, and a family should be supported in making the appropriate choice for their family, if deemed appropriate by the metabolic healthcare team.
A specialist metabolic team should be responsible for the management of the individual with Classical homocystinuria (HCU) from diagnosis and throughout life.
For more information and guidance see the resource below.

- Morris AAM, Kožich V, Santra S, Andria G, Ben-Omran TIM, Chakrapani AB, et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017;40:49–74. https://doi.org/10.1007/s10545-016-9979-0
- Dixon M, MacDonald A, White F. Disorders of amino acid metabolism, organic acidaemias and urea cycle disorders. In: Shaw V, editor. Clinical paediatric dietetics. 5th ed. Hoboken (NJ): Wiley-Blackwell; 2020. p. 513–98.
- Andria G, Fowler B, Sebastio G. Disorders of sulfur amino acid metabolism. In: Saudubray J-M, van den Berghe G, Walter JH, editors. Inborn metabolic diseases: diagnosis and treatment. 5th ed. Berlin: Springer; 2012. p. 311–21.
- Adam S, Almeida MF, Weber EC, Champion H, Chan H, Daly A, et al. Dietary practices in pyridoxine non-responsive homocystinuria: a European survey. Mol Genet Metab. 2013;110:454–9. https://doi.org/10.1016/j.ymgme.2013.10.003
- Hoss GRW, Sperb-Ludwig F, Schwartz IVD, Blom HJ. Homocystinuria: a common inborn error of metabolism? An epidemiological study based on genetic databases. Mol Genet Genomic Med. 2020;8:e1214. https://doi.org/10.1002/mgg3.1214
Related products
Browse the products available for managing this condition.


Information presented on this page is intended for healthcare professional use only. Food for Special Medical Purposes to be used under medical supervision.
Date of publication: 28/05/2025
