Phenylketonuria, or PKU, is an inherited metabolic disorder caused by the defect in the enzyme (phenylalanine hydroxylase) responsible for converting amino acid phenylalanine to tyrosine. The body cannot get rid of the excess amounts of phenylalanine (Phe), it builds up in the blood and other body tissues, and some is converted to substances called phenylketones. The high level of phenylalanine, phenylketones, and low tyrosine levels result in poor growth, neurological damage, intellectual disability and epilepsy.
In genetic terms, PKU is an autosomal recessive disorder. This means that an affected person has two PKU genes – one from each parent.
PKU affects between 1 in 10,000 and 1 in 20,000 people depending on the country of origin. In some countries, the prevalence is higher. In Turkey, as an example, the prevalence reaches 1 PKU patient in 2600 newborns.
PKU is identified through newborn screening. PKU screening is mandatory for all newborns in many countries. Early detection and treatment is extremely important. Without treatment, the person could become severely retarded. With careful and prompt treatment, the individuals with PKU can develop normally and live healthy lives.
PKU is treated with a special diet involving protein restriction and supplementation with a special formula. This treatment allows to keep the blood Phe levels down to a safe level.
Foods and drinks that contain high levels of Phe, should be avoided. Foods and drinks that contain lower levels of Phe are allowed in measured amounts.
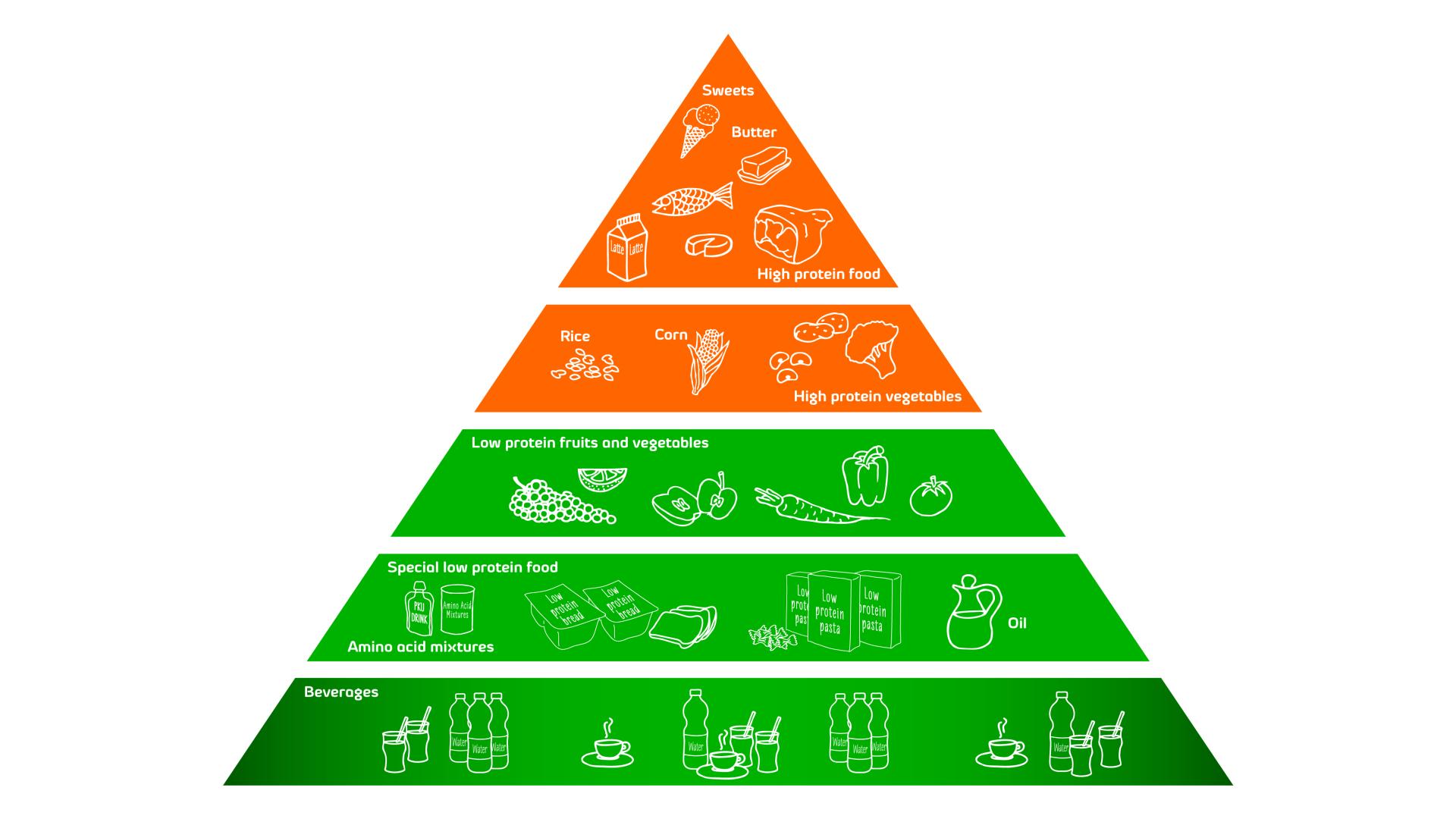
Since children and especially babies, need protein in order to grow and develop normally, special formulas have been developed to replace the high protein foods. The first of those products which Comidamed offers is a milk substitute called comida-PKU A formula. This is a special formula, free from Phenylalanine, designed especially for infants in their first year of life. In the dietary management of children from 1 year of age, adolescents and adults comida-PKU B formula and comida-PKU B can be used. comida-PKU C is a phenylalanine free, concentrated protein supplement for adolescents from 15 years of age and adults.

